Molekülleri süperbilgisayarsız çözmenin yeni yolu: XC’de makine öğrenimi
Kuantum çok-gövdeli hesaplardan ‘tersine’ öğrenerek ‘Yoğunluk Fonksiyonel Teorisi’nin kalbi olan değişim-korelasyon (XC) fonksiyoneline yeni bir yaklaşım geliştirildi. Yöntem pil malzemelerinden ilaç keşfine, yarı iletkenlerden kuantum hesaplamaya uzanan geniş bir alanda simülasyon maliyetini düşürmeyi hedefliyor.

Malzemelerin ve kimyasal reaksiyonların atom-altı davranışını yakalamanın altın standardı, tek tek elektronları izleyen kuantum çok-gövdeli yöntemler. Ancak bu yöntemlerin maliyeti o kadar yüksek ki, ABD ulusal laboratuvarlarındaki süperbilgisayar kaynaklarının üçte birini tüketiyor ve pratikte yalnızca birkaç elektronlu sistemlerle sınırlı kalıyor. Bu darboğazı aşmak için kullanılan Yoğunluk Fonksiyonel Teorisi (DFT) ise elektronu tek tek değil, elektron yoğunluğu üzerinden ele alarak yüzlerce atomu makul sürede simüle edebiliyor; fakat DFT’nin doğruluğu, çekirdeğinde yer alan değişim-korelasyon (XC) fonksiyonelinin ne kadar iyi modellendiğine bağlı.
KAYIP DENKLEME ATAK
Araştırma ekibi, DFT’nin “evrensel” XC fonksiyoneline bir adım daha yaklaşmak için problemi tersine çevirdi. Michigan Üniversitesi’nden Prof. Vikram Gavini’nin sorumlu yazar olduğu çalışmada, önce tek atomlar ve küçük moleküller kuantum çok-gövdeli yöntemlerle yüksek doğrulukta çözüldü; ardından makine öğrenmesi ile “bu doğru elektron davranışını hangi XC fonksiyoneli verir?” sorusunun yanıtı arandı. Kimya profesörü Paul Zimmerman’ın ekibi, lityum, karbon, azot, oksijen, neon ile H₂ ve LiH’ten oluşan bir eğitim veri seti oluşturdu; flor ve su gibi eklemeler ise modeli daha da iyileştirmedi ve hafif türlerden alınan bilginin doygunlaştığı görüldü.
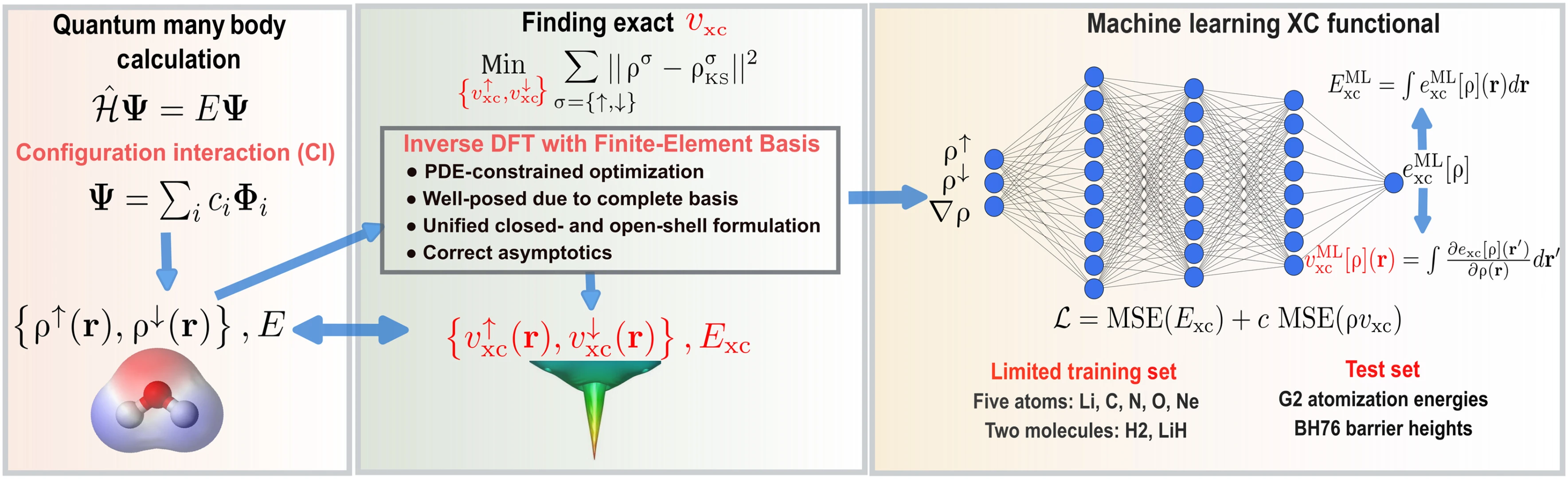
İKİNCİ BASAMAKLA ÜÇÜNCÜ DOĞRULUK
DFT’de doğruluk genellikle bir ‘merdiven’ olarak betimleniyor: En altta yerel yaklaşımlar, bir üstte gradyan bilgisi katan GGA (ikinci basamak), onun üzerinde ise kinetik enerji yoğunluğu gibi ek değişkenlere dayanan meta-GGA (üçüncü basamak) bulunuyor. Ekip, yalnızca ikinci basamak karmaşıklığında kalarak üçüncü basamak seviyesine yaklaşan sonuçlar elde etti. Bu, daha karmaşık dalga fonksiyonu türevleri olmadan yüksek doğruluk sağlayan yeni bir XC fonksiyonelinin mümkün olduğuna işaret ediyor.
MAKİNE ÖĞRENİMLİ FONKSİYONEL
Araştırmacılar, kuantum çok-gövdeli “doğru cevapları” kompakt ve hızlı bir forma dökerek, DFT’nin en pahalı halkasını zayıf nokta olmaktan çıkarmayı amaçlıyor. Ekibin ilk yazarı Dr. Bikash Kanungo, malzemeye bağımlı olmayan, genel amaçlı bir XC fonksiyonelinin, pil malzemeleri, ilaç keşfi, yarı iletken tasarımı ve kuantum bilgi işleme gibi alanlarda yaygın yeniden kullanım sağlayabileceğini vurguluyor.

HESAPLANABİLİRLİK VE YOL HARİTASI
Enerji Bakanlığı desteğiyle yürütülen projede, ABD’nin en büyük süperbilgisayarlarında yapılan deneyler, yoğunluk gradyanları ile sınırlı yaklaşımda bile ciddi kaynak gerektirdi. Bir sonraki adımda ekip, katı malzemeler için aynı tersine-öğrenme stratejisini deneyecek:
• Aynı fonksiyonel katılarda da çalışacak mı?
• Katılar için ayrı optimize edilen bir fonksiyonel daha iyi sonuç verir mi?
• Molekül-katı ayrımını aşan birleşik bir fonksiyonel mümkün mü?
ORBİTALLERE DOĞRU
Daha da yüksek doğruluk için, yalnızca yoğunluklara değil, orbitallere dayalı bilgileri de içeren şemalara geçmek gerekecek. Bu, tersine çözümün matematiğini zorlaştırsa da doğru XC’ye giden yolu netleştirebilir. Ekip, daha fazla hesaplama zamanı ve genişleyen veri setleriyle, ‘evrensel’ fonksiyonelin formunu aydınlatmayı hedefliyor.

MALZEME KEŞFİNDE HIZ
Yeni yaklaşım, hesaplama maliyetini düşürürken öngörü gücünü yükseltmeyi vaat ediyor. Daha iyi pil elektrotları, daha etkili katalizör yüzeyleri, daha hassas ilaç-hedef etkileşimleri ve kuantum malzemelerinin tasarımında, DFT’nin güven sınırlarını genişleterek tarama (screening) çalışmalarını hızlandırabilir.

Meta, sürekli kayıt alabilen 'süper algılama'lı yeni akıllı gözlükleri test ediyor

Avustralyalı ABC, haber üretiminde Claude yapay zekasını kullanacak

Yapay zeka iki yeni süperiletken malzemenin keşfini hızlandırdı

SpaceX, son altı ayda 260 Starlink uydusunu imha etti

Hayabusa2, 100 milyon km uzaktaki Torifune asteroitinin yakınından geçti

MeerKAT teleskobu 'Mavi Gözlü Pulsar'dan radyo sinyalleri yakaladı

Otonom sistemler için dünyanın ilk sertifikalı 3D ultrasonik sensörü tanıtıldı

Çin, ABD ambargolarını aşmak için 3D yığınlama teknolojisine yatırım yapıyor
Yorum yazmak için giriş yapın.
Yorumlar yükleniyor…